La neurodegeneración asociada a la proteína de membrana mitocondrial es causada por mutaciones en el gen C19orf12 . Este gen se encuentra en el cromosoma 19 y se cree que desempeña un papel en el metabolismo de los ácidos grasos. Aunque es muy poco frecuente, es una de las formas más comunes de neurodegeneración asociada a la proteína de membrana mitocondrial. La función de la proteína C19orf12 sigue siendo desconocida, pero puede estar involucrada en la función mitocondrial, la homeostasis lipídica y el metabolismo de la coenzima A.
MPAN es una de las principales formas de NBIA y presenta síntomas clínicos distintivos que la diferencian de otras formas de NBIA.
El inicio suele ocurrir en la infancia (entre los 3 y los 16 años, lo que se considera inicio juvenil) o en la adultez temprana (entre los 17 y los 24 años, lo que se considera inicio en la edad adulta).
Sintomas
MPAN se caracteriza con espasticidad más prominente que la distonía, debilidad en los músculos causada por neuropatía axonal motora, atrofia óptica bilateral. Los signos tempranos de motoneurona superior (signos piramidales, por ejemplo, espasticidad) son hallazgos constantes, seguidos posteriormente por signos de disfunción de la motoneurona inferior (pérdida de los reflejos tendinosos profundos, debilidad muscular y atrofia). Más de la mitad de los pacientes presenta distonía progresiva, parkinsonismo. Son comunes la pérdida de peso y la disfunción urinaria y/o intestinal. Marcha idiopatica ( caminar de puntillas).
La mayoría de las personas afectadas aún pueden caminar cuando llegan a la edad adulta. Los signos psiquiátricos son comunes, incluyendo comportamiento impulsivo o compulsivo, depresión y cambios de humor frecuentes. A diferencia de la mayoría de las otras formas de NBIA, la gran mayoría de las personas con MPAN desarrollan un deterioro cognitivo progresivo.
Diagnostico
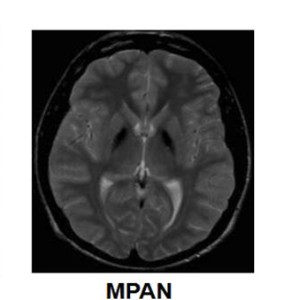
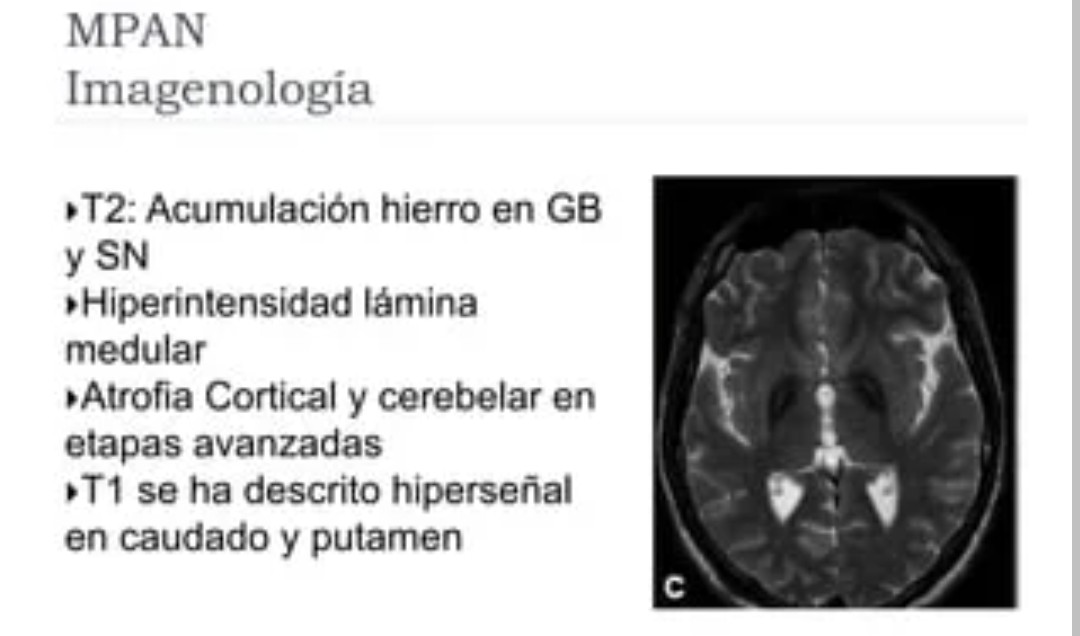
El diagnóstico se basa en la neuroimagen que pone en evidencia los depósitos de hierro, principalmente en el globo pálido y en la sustancia negra, a menudo con una estriación hiperintensa característica en T2 entre el globo pálido interno hipointenso y el globo pálido externo. Los exámenes oftalmológicos y los potenciales evocados visuales son importantes para identificar la atrofia óptica. El examen neuropatológico muestra esferoides axonales, cuerpos de Lewy e inclusiones que contienen tau hiperfosforilada. El estudios genetico de mutaciones en el gen C19orf12 confirma el diagnóstico.
Tratamiento
No existe cura se recomienda un equipo de profesionales médicos que recomiende tratamientos según los síntomas actuales.
Las estrategias de abordaje terapéutico se centran en el cuidado paliativo médico o quirúrgico de los síntomas.
Los médicos también pueden intentar controlar la enfermedad realizando exámenes oculares periódicos, así como pruebas neurológicas para detectar distonía, espasticidad y parkinsonismo. Esto puede incluir la evaluación de la capacidad de la persona afectada para caminar y hablar, para una posible terapia física, ocupacional y/o del habla. Los síntomas neuropsiquiátricos pueden requerir tratamiento por parte de un psiquiatra.
Terapias para manejar la distonía
Toxina botulínica intramuscular: el bótox se inyecta en los músculos espásticos y distónicos para ayudarlos a relajarse durante un período de tiempo.
Artane (trihexifenidilo), tomado por vía oral, generalmente dividido en múltiples dosis cada día
Baclofeno (oral o intratecal): Uno de los principales fármacos utilizados para tratar la distonía, que suele administrarse primero por vía oral y dividirse en varias dosis al día. En el método intratecal, una bomba de baclofeno implantada administra el medicamento directamente al líquido cefalorraquídeo.
Estimulación cerebral profunda: se utiliza cada vez con más frecuencia en la NBIA y hay algunas evidencias de que es beneficiosa. Un estimulador envía impulsos eléctricos a la región cerebral afectada para ayudar a que los músculos se relajen. Implica la implantación quirúrgica de un electrodo, una extensión y un paquete de baterías (IPG). El electrodo contiene 4 electrodos y se implanta en la región del globo pálido del cerebro. La extensión conecta el electrodo al paquete de baterías (IPG). El IPG es un neuroestimulador alimentado por batería que se coloca en el abdomen (o en algunos casos debajo de la clavícula)
Fisioterapia y terapia ocupacional. Puede estar indicada o no para quienes presentan síntomas leves.
Genetica
La MPAN es un trastorno hereditario y, en la mayoría de los casos, autosómico recesivo. Las enfermedades recesivas solo se producen cuando ambos padres son portadores de la misma enfermedad y luego transmiten sus genes modificados a su hijo, lo que da lugar a dos mutaciones causantes de la enfermedad.
La MPAN también se hereda en casos raros como trastorno autosómico dominante. Los individuos afectados por el gen mutado dominante tienen solo una mutación en uno de sus cromosomas. Se los llama portadores dominantes y si la mutación dominante pasa a su hijo, este se verá afectado. Neurodegeneración asociada a la proteína de membrana mitocondrial autosómica dominante (MPAN).
Pronostico
La progresión de la MPAN suele ser lenta y la supervivencia se mantiene hasta bien entrada la edad adulta. Sin embargo, se han descrito casos raros de pacientes con inicio repentino en la edad adulta y progresión rápida. Los pacientes con MPAN aprenden a caminar y suelen ser móviles hasta principios de la edad adulta.
Toda la información es solo para fines informativos. No respaldamos estudios específicos ni ensayos clínicos, medicamentos experimentales, procedimientos ni empresas biotecnológicas o farmacéuticas.
Busque en ClinicalTrials.gov en los EE. UU. y en el Registro de ensayos clínicos de la UE en Europa para obtener información sobre estudios clínicos.
